关于TCGAbiolinks包的学习前面一共介绍了5篇推文。
今天继续学习如何使用TCGAbiolinks下载和整理MAF格式的突变数据。
之前的TCGA的MAF文件是可以下载的,每个癌症包含4种软件得到的突变文件:

后来就改版了,不让你随便下载了。但其实还是可以下载的,只不过没有那么多选择了!
现在的情况是每个样本都是一个单独的maf文件,需要下载后自己整理,就像整理表达矩阵那样。
MAF文件的下载
但是现在我们有TCGAbiolinks,根本不需要自己动手,直接三步走即可得到我们需要的MAF文件。
library(TCGAbiolinks)
query <- GDCquery(
project = "TCGA-COAD",
data.category = "Simple Nucleotide Variation",
data.type = "Masked Somatic Mutation",
access = "open"
)
GDCdownload(query)
GDCprepare(query, save = T,save.filename = "TCGA-COAD_SNP.Rdata")
这样得到的这个Rdata文件其实是一个数据框,不过由于内容和之前的MAF文件一模一样,所以也是可以直接用maftools读取使用的。
maftools是一个非常强大的突变数据可视化和分析的R包,这个包在bioconductor上,需要的自行安装。
无缝对接maftools
由于我们在第一步已经下载过了,所以这里就不用下载了,直接加载保存好的数据。
我们以TCGA-COAD的数据作为演示。
library(maftools)
load(file = "./TCGA-SNP/TCGA-COAD_SNP.Rdata")
maf.coad <- data
简单看一下这个数据:
class(maf.coad)
## [1] "data.frame"
dim(maf.coad)
## [1] 252664 141
maf.coad[1:10,1:10]
## X1 Hugo_Symbol Entrez_Gene_Id Center NCBI_Build Chromosome Start_Position
## 1 1 AGRN 375790 BCM GRCh38 chr1 1046481
## 2 1 ACAP3 116983 BCM GRCh38 chr1 1295539
## 3 1 CALML6 163688 BCM GRCh38 chr1 1916980
## 4 1 PRKCZ 5590 BCM GRCh38 chr1 2150972
## 5 1 WRAP73 49856 BCM GRCh38 chr1 3635995
## 6 1 CHD5 26038 BCM GRCh38 chr1 6142440
## 7 1 CAMTA1 23261 BCM GRCh38 chr1 7663513
## 8 1 ERRFI1 54206 BCM GRCh38 chr1 8014193
## 9 1 SLC2A7 155184 BCM GRCh38 chr1 9022922
## 10 1 PGD 5226 BCM GRCh38 chr1 10411462
## End_Position Strand Variant_Classification
## 1 1046481 + Frame_Shift_Del
## 2 1295539 + Missense_Mutation
## 3 1916980 + Silent
## 4 2150972 + Silent
## 5 3635995 + Silent
## 6 6142440 + Missense_Mutation
## 7 7663513 + Silent
## 8 8014193 + Missense_Mutation
## 9 9022922 + Missense_Mutation
## 10 10411462 + Silent
可以看到是一个data.frame类型的文件。
这个文件一共有252664行,141列,包含了gene symbol,突变类型,突变位置,导致的氨基酸变化等信息。
下面就直接用read.maf()函数读取即可,没有任何花里胡哨的操作!
maf <- read.maf(maf.coad)
## -Validating
## -Silent variants: 63597
## -Summarizing
## --Mutiple centers found
## BCM;WUGSC;BCM;WUGSC;BCM;BI--Possible FLAGS among top ten genes:
## TTN
## SYNE1
## MUC16
## -Processing clinical data
## --Missing clinical data
## -Finished in 6.000s elapsed (5.690s cpu)
然后就是进行各种可视化操作,毫无难度:
plotmafSummary(maf = maf, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE)

是不是非常简单,虽然没有直接提供单个癌症的MAF文件,但是使用TCGAbiolinks后,会直接帮我们整理好,没有任何难度。
如果你由于各种原因不能使用这个包下载数据,那你可以直接用网页下载,然后按照我之前的推文进行整理:
xxxxxxxxxxxxx
但是这个方法用在表达谱数据是没有问题的,理论上用在其他类型的数据都是可以的,但是我并没有尝试过,欢迎大家使用后留言。
如果你在网络上看见一个叫xxx.pl的文件,并且需要付费获取,建议你不要花这个冤枉钱,不值那个价,希望大家多多擦亮眼睛!
如果你非要用手撕代码的方式自己整理,也是非常简单的,比整理转录组数据的表达矩阵简单100倍。
自己整理成MAF格式
首先你要去GDC TCGA的官网下载某个癌症的所有的maf文件,还是以TCGA-COAD为例,下载好之后是这样的:
每个样本第一个文件夹,每个文件夹下面有一个.gz结尾的压缩文件,这个文件解压缩之后就是大家熟悉的.maf文件大,但是只是一个样本的~

把这个.maf文件用VScode打开后是这样的:
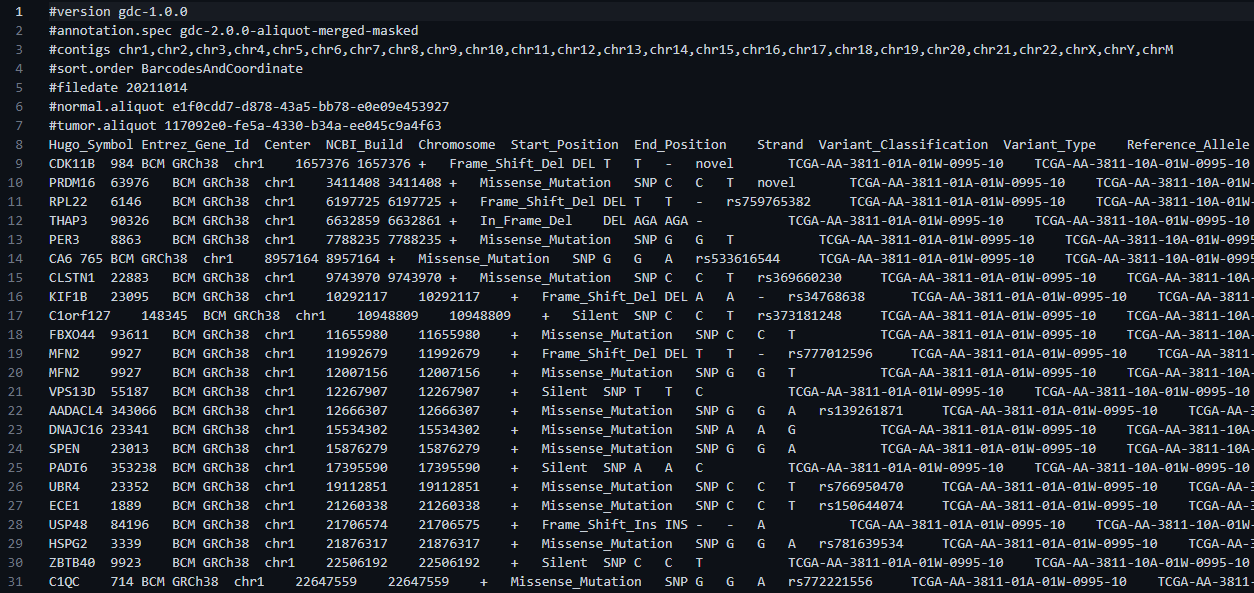
不妨多解压几个打开看一看,都是一样的结构,所以就很简单了,把所有的文件读取进来然后直接rbind()即可。
但是在此之前我们可以先读取一个试试看:
# 路径必须正确
tmp <- read.table("G:/tcga/GDCdata/TCGA-COAD/harmonized/Simple_Nucleotide_Variation/Masked_Somatic_Mutation/007c2ae4-bbd2-42c6-ab67-bf016fbddb51/982004b5-52e1-4a69-97d3-25bdcb77b026.wxs.aliquot_ensemble_masked.maf.gz",
skip = 7, # 前面7行都不要
sep = "\t", # 必须指定
header = T # 有行名
)
tmp[1:10,1:8]
## Hugo_Symbol Entrez_Gene_Id Center NCBI_Build Chromosome Start_Position
## 1 NOC2L 26155 BCM GRCh38 chr1 945088
## 2 SDF4 51150 BCM GRCh38 chr1 1217753
## 3 B3GALT6 126792 BCM GRCh38 chr1 1233752
## 4 MIB2 142678 BCM GRCh38 chr1 1629520
## 5 NADK 65220 BCM GRCh38 chr1 1765325
## 6 GNB1 2782 BCM GRCh38 chr1 1804562
## 7 PANK4 55229 BCM GRCh38 chr1 2510063
## 8 PRXL2B 127281 BCM GRCh38 chr1 2588386
## 9 PRDM16 63976 BCM GRCh38 chr1 3412316
## 10 WRAP73 49856 BCM GRCh38 chr1 3633442
## End_Position Strand
## 1 945088 +
## 2 1217753 +
## 3 1233752 +
## 4 1629520 +
## 5 1765325 +
## 6 1804562 +
## 7 2510063 +
## 8 2588386 +
## 9 3412316 +
## 10 3633442 +
非常顺利,和上面那个整理好的格式一模一样,唯一不同就是这个只是一个样本的。
下面我们就批量读取并合并就好了!
# 确定文件路径!
dir.path <- "G:/tcga/GDCdata/TCGA-COAD/harmonized/Simple_Nucleotide_Variation/Masked_Somatic_Mutation"
# 获取所有maf文件路径
all.maf <- list.files(path = dir.path, pattern = ".gz",
full.names = T, recursive = T)
# 看看前3个
all.maf[1:3]
## [1] "G:/tcga/GDCdata/TCGA-COAD/harmonized/Simple_Nucleotide_Variation/Masked_Somatic_Mutation/007c2ae4-bbd2-42c6-ab67-bf016fbddb51/982004b5-52e1-4a69-97d3-25bdcb77b026.wxs.aliquot_ensemble_masked.maf.gz"
## [2] "G:/tcga/GDCdata/TCGA-COAD/harmonized/Simple_Nucleotide_Variation/Masked_Somatic_Mutation/010f9040-294d-4d14-a2b4-80d7a11625dd/5083b949-1bf3-4bc2-bf4f-f668f8a13792.wxs.aliquot_ensemble_masked.maf.gz"
## [3] "G:/tcga/GDCdata/TCGA-COAD/harmonized/Simple_Nucleotide_Variation/Masked_Somatic_Mutation/0148fff1-b8af-4bf0-8bcd-de1ff9f750f3/2c16cfe2-bf6d-4a39-af3a-9dfd5ada3e17.wxs.aliquot_ensemble_masked.maf.gz"
然后直接读取就行了,觉得慢可以用data.table::fread()加快速度。
maf.list <- lapply(all.maf, read.table,
skip = 7,
sep = "\t",
header = T)
然后直接合并即可,如果不放心可以看看列数列名是不是一样,100%一样,我们就不看了。
# lapply(maf.list, dim)
maf.merge <- do.call(rbind,maf.list)
目前为止看似一切顺利,本以为即将结束,但是没想到横生枝节!
竟然读取不了,而且我们得到的这个maf.merge竟然只有137665行!和252664行的差距实在是太大了!
# 读取失败!
maf1 <- read.maf(maf.merge)
## -Validating
## --Removed 5 duplicated variants
## --Non MAF specific values in Variant_Classification column:
## 3UTR DEL T T - novel TCGA-A6-6781-01A-22D-1924-10 TCGA-A6-6781-10A-01D-1924-10
果然不检查数据是不行的!
然后只能回过头去看哪里出了问题,通过仔细使用VScode直接打开maf文件和我们读取的文件对比,发现了问题。
在Variant_Classification这一列中,有一些3'UTR / 5'UTR这样的类型,但是在使用read.table()读取的时候竟然识别不出来!
小丑竟是我自己!
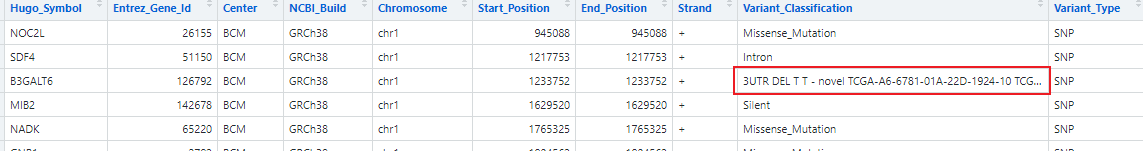
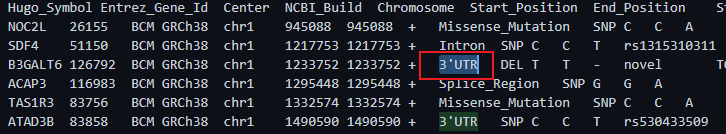
所以直接导致遇到这个之后的所有行都是错位的,而且少了非常多行。
生气啊!
但是找到问题之后解决就非常简单,换个函数就行了,我们直接用data.table::fread()读取!
maf.list <- lapply(all.maf, data.table::fread,
sep = "\t",
header = T,
skip = 7
)
maf.merge <- do.call(rbind,maf.list)
dim(maf.merge)
## [1] 252664 140
现在就和前面的数据一模一样了,252664行,140列(少了一列是表示来自于第几个样本,没有用)。
# 读取成功!
maf1 <- read.maf(maf.merge)
## -Validating
## -Silent variants: 63597
## -Summarizing
## --Mutiple centers found
## BCM;WUGSC;BCM;BI;BCM;WUGSC--Possible FLAGS among top ten genes:
## TTN
## SYNE1
## MUC16
## -Processing clinical data
## --Missing clinical data
## -Finished in 3.970s elapsed (3.730s cpu)
plotmafSummary(maf = maf1, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE)
简单!下次说说这个maftools的使用。
觉得有用请多多转发~拒绝不必要的花钱!难道免费的不如付费的香??