写在前面——经过前面的一系列分析,我们得到了几个counts数据,接下来就需要根据这些数据来进行分析。本文使用Rstudio,从序列比对结果中筛选出差异基因,目的是(根据不同基因的表达量)找出实验组与对照组的差异。
本文使用的数据见RNA-seq——上游分析练习(数据下载+hisat2+samtools+htseq-count)
参考:
RNA-seq(6): reads计数,合并矩阵并进行注释
RNA-seq(7): DEseq2筛选差异表达基因并注释(bioMart)
1. 合并矩阵并进行注释
rm(list = ls())
options(stringsAsFactors = FALSE)
# 读取数据
control_1 <- read.table("SRR3589959.count", col.names = c("gene_id", "control_1"))
control_2 <- read.table("SRR3589961.count", col.names = c("gene_id", "control_2"))
treat_1 <- read.table("SRR3589960.count", col.names = c("gene_id", "treat_1"))
treat_2 <- read.table("SRR3589962.count", col.names = c("gene_id", "treat_2"))
# 将数据合并
raw_count <- merge(merge(control_1, control_2, by = "gene_id"),
merge(treat_1, treat_2, by = "gene_id"))
# head(raw_count)
# 去除无用信息
raw_count_filt <- raw_count[-1:-5,]
# 修改行名
ENSEMBL <- gsub("\\.\\d*", "", raw_count_filt$gene_id)
row.names(raw_count_filt) <- ENSEMBL
处理完的raw_count_filt:
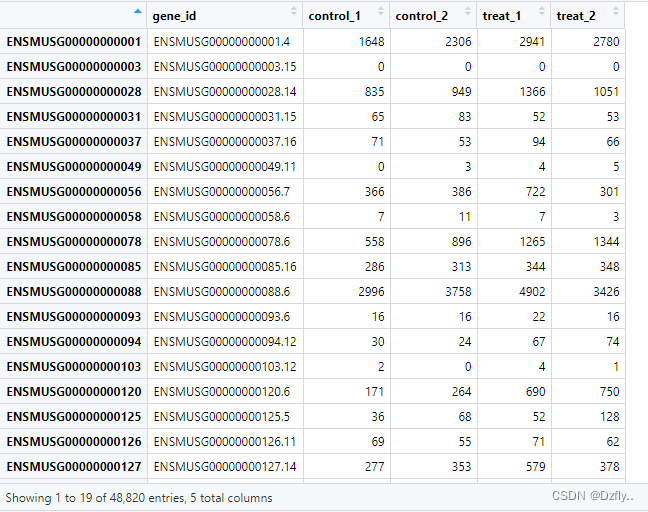
# 将ensembl_gene_id转化为gene_symbol
library(biomaRt)
library(curl)
my_ensembl_gene_id <- row.names(raw_count_filt)
mart <- useDataset("mmusculus_gene_ensembl", useMart("ensembl"))
mms_symbols <- getBM(attributes = c("ensembl_gene_id", "external_gene_name",
"description"),
filters = "ensembl_gene_id", values = my_ensembl_gene_id,
mart = mart)
# 方便按照ensembl_gene_id来合并两个数据集
raw_count_filt <- cbind(ENSEMBL, raw_count_filt)
colnames(raw_count_filt)[1] <- c("ensembl_gene_id")
# 合并
readcount <- merge(raw_count_filt, mms_symbols, by = "ensembl_gene_id")
# 保存
write.csv(readcount, file = "readcount.csv")
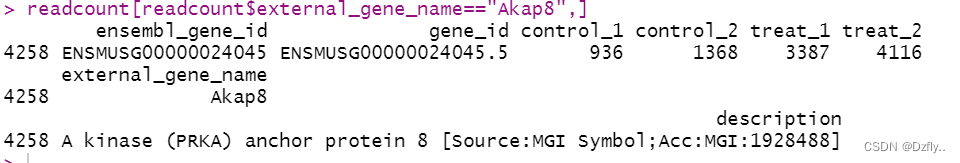
# 查看Akap8的表达情况
readcount[readcount$external_gene_name=="Akap8",]
# 整理数据,方便后续使用
rownames(readcount) <- readcount$ensembl_gene_id
mycounts <- readcount[,3:6]
write.csv(mycounts, file = "mycounts.csv")
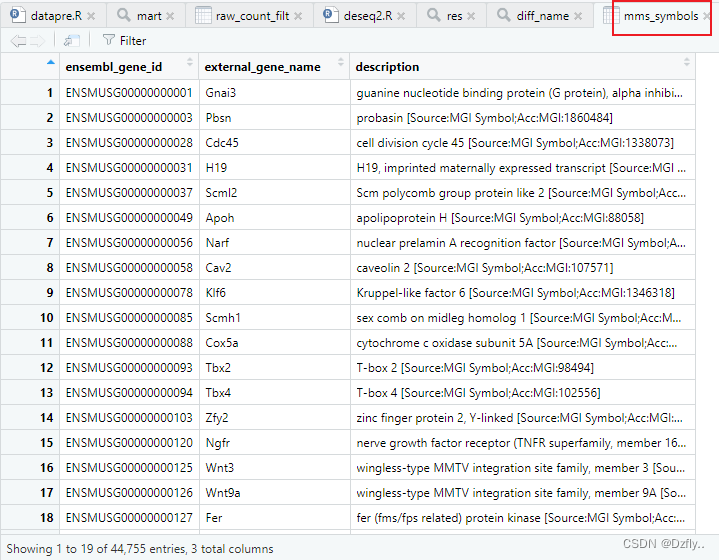

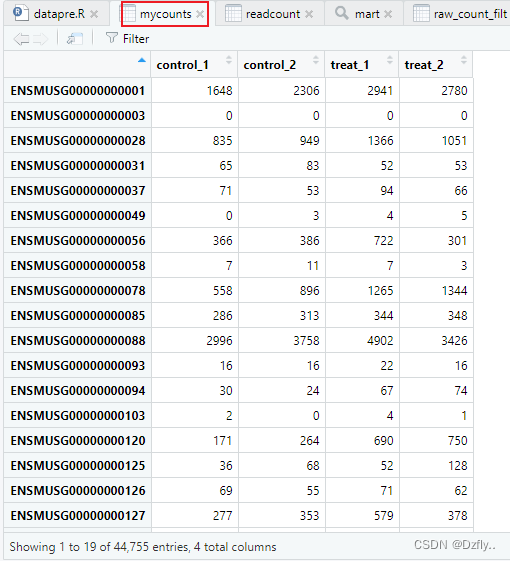
整理好的数据包含Ensembl ID以及每组的基因比对结果。
2. 筛选差异基因(DESeq2)
rm(list = ls())
library(tidyverse)
library(DESeq2)
# 读取上一步整理好的数据
mycounts <- read.csv("mycounts.csv")
rownames(mycounts) <- mycounts$X
mycounts <- mycounts[,-1]
# 设置factor
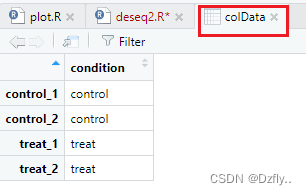
condition <- factor(c(rep("control", 2), rep("treat", 2)), levels = c("control", "treat"))
# 生成colData
colData <- data.frame(row.names = colnames(mycounts), condition)
# 使用DESeq2分析
dds <- DESeqDataSetFromMatrix(mycounts, colData, design = ~ condition)
dds <- DESeq(dds)
res <- results(dds, contrast = c("condition", "control", "treat"))
res <- res[order(res$pvalue),]
# summary(res)
# table(res$padj<0.05)
write.csv(res, file = "all_results.csv")
# 筛选差异基因
# P和log2FC的值可以自己设置,值不同,筛选出来的差异基因数目也不同
diff_gene_deseq2 <- subset(res, padj < 0.05 & abs(log2FoldChange) >1)
write.csv(diff_gene_deseq2, file = "DEG_treat_vs_control.csv")
# 将Ensembl ID转化为Gene Symbol
# 方法一
# library(clusterProfiler)
# library(org.Mm.eg.db)
#
# name <- bitr(rownames(diff_gene_deseq2), fromType = "ENSEMBL", toType = "SYMBOL",
# OrgDb = "org.Mm.eg.db")
# 方法二
library(biomaRt)
library(curl)
my_ensembl_gene_id <- row.names(diff_gene_deseq2)
mart <- useDataset("mmusculus_gene_ensembl", useMart("ensembl"))
mms_symbols <- getBM(attributes = c("ensembl_gene_id", "external_gene_name",
"description"),
filters = "ensembl_gene_id", values = my_ensembl_gene_id,
mart = mart)
ensembl_gene_id <- rownames(diff_gene_deseq2)
diff_gene_deseq2 <- cbind(ensembl_gene_id, diff_gene_deseq2)
colnames(diff_gene_deseq2)[1] <- c("ensembl_gene_id")
# 得到最终结果
diff_name <- merge(diff_gene_deseq2, mms_symbols, by = "ensembl_gene_id")
diff_name[diff_name$external_gene_name=="Akap8",]

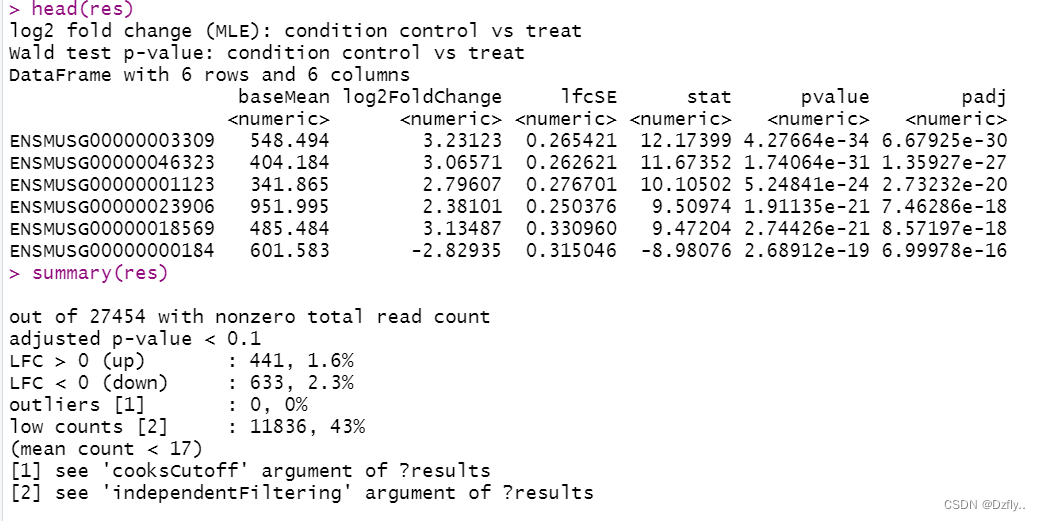

至此,我们便得到了差异基因,之后就可以根据这些差异基因,进行基因的富集分析,可以更加细致的了解到实验组的变化。更重要的是,之后可以作出一些精美的图片,帮助我们发文章~~/(ㄒoㄒ)/~~