时隔一年多,终于又回来写记录文章啦~
现在要自学生信分析,在网上找了一个不错的教程。生信入门肯定要学Linux,所以按照教程,做了Linux必做20题。下面是详细的答案,花费了两三天时间完成的,觉得不错的话就点个赞吧~
视频教程:https://www.bilibili.com/video/BV1ds411g7eg?p=5&spm_id_from=pageDriver
题目原文:http://www.bio-info-trainee.com/2900.html
推荐网站:https://man.linuxde.net/ https://wiki.jikexueyuan.com/
推荐书籍:Linux命令行与shell脚本编程大全
1. 在任意文件夹下面创建形如 1/2/3/4/5/6/7/8/9 格式的文件夹系列。
mkdir -p 1/2/3/4/5/6/7/8/9
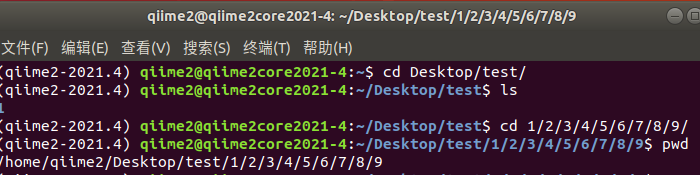
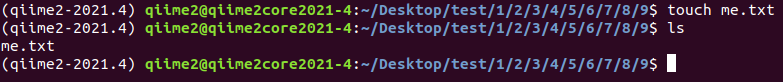
2. 在创建好的文件夹(/home/qiime2/Desktop/test/1/2/3/4/5/6/7/8/9)下创建文本文件 me.txt
touch me.txt
3. 在文本文件 me.txt 里面输入内容:
Go to: http://www.biotrainee.com/
I love bioinfomatics.
And you ?
cat > me.txt
Go to: http://www.biotrainee.com/
I love bioinfomatics.
And you ?
# ctrl+c 停止输入并退出
cat me.txt # 查看文件内容

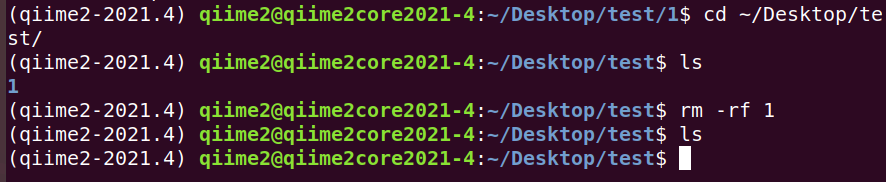
4. 删除上面创建的文件夹 1/2/3/4/5/6/7/8/9 及文本文件 me.txt
# 先退回到初始文件夹1的上一层,然后删除1,一步到位
rm -rf 1
5. 在任意文件夹下面创建 folder_1~5这5个文件夹,然后每个文件夹下面再创建 folder_1~5这5个文件夹
mkdir floder_{1..5}
mkdir floder_{1..5}/floder_{1..5}

亦可一步完成
mkdir -p floder_{1..5}/floder_{1..5}

6. 在第5题创建的每一个文件夹下面都创建一个文本文件 me.txt ,文本内容同第2题。
# 使用脚本来完成
vim txt.sh
# 脚本内容
for I in {1..5};do
cd ~/Desktop/test/floder_$I
for i in {1..5};do
cd ~/Desktop/test/floder_$I/floder_$i
echo -e "Go to: http://www.biotrainee.com/ \nI love bioinfomatics. \nAnd you ?" > me.txt
done
done
# 运行脚本
bash txt.sh

7. 再次删除掉前面几个步骤建立的文件夹及文件
# *表示所有前缀都是floder_的文件
rm -r floder_*

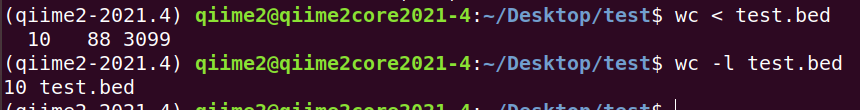
8. 下载 http://www.biotrainee.com/jmzeng/igv/test.bed 文件,查看含有 H3K4me3 的那一行是第几行?该文件总共有几行?
# -c 断点续传
wget -c http://www.biotrainee.com/jmzeng/igv/test.bed

grep -n H3K4me3 test.bed

# 输出 行数、词数、字节数
wc < test.bed
# 只显示行数
wc -l test.bed
9. 下载 http://www.biotrainee.com/jmzeng/rmDuplicate.zip 文件,并且解压,查看里面的文件夹结构
wget -c http://www.biotrainee.com/jmzeng/rmDuplicate.zip
unzip rmDuplicate.zip
tree rmDuplicate


10. 打开第9题解压的文件,进入 rmDuplicate/samtools/single 文件夹里面,查看后缀为 .sam 的文件,搞清楚生物信息学里面的SAM/BAM定义是什么。
cd rmDuplicate/samtools/single/
# 查看后缀为sam的文件
less tmp.sam
# 查看该文件的前五行 (head默认为前十行)
cat tmp.sam | head -5

SAM和BAM的介绍:https://www.omicsclass.com/article/241
当我们测序得到的fastq数据map到基因组之后,会得到一个以sam或bam为扩展名的文件。这里,SAM的全称是sequence alignment/map format。而BAM就是SAM的二进制文件,也就是压缩格式的sam文件。
由于sam格式的文件通常都非常大,所以为了节省存储空间而将sam转换为二进制格式以便于存储,也就是bam文件。 sam/bam文件可以由特定的一些软件(比如samtools)来处理的,包括格式互转、排序、建立索引等操作。
11. 安装 samtools 软件
# 下载安装包
wget -c https://sourceforge.net/projects/samtools/files/samtools/1.12/samtools-1.12.tar.bz2
# 解压
tar -xjvf samtools-1.12.tar.bz2
# 编译
##参考##
# https://www.cnblogs.com/tinywan/p/7230039.html
# https://blog.csdn.net/cuicanlianyue/article/details/79458594
cd samtools-1.12/
make
## Linux中常见的解压缩命令
# 1、*.tar 用 tar –xvf 解压
# 2、*.gz 用 gzip -d或者gunzip 解压
# 3、*.tar.gz和*.tgz 用 tar –xzvf 解压
# 4、*.bz2 用 bzip2 -d或者用bunzip2 解压
# 5、*.tar.bz2用tar –xjvf 解压
# 6、*.Z 用 uncompress 解压
# 7、*.tar.Z 用tar –xZvf 解压
# 8、*.rar 用 unrar e解压
# 9、*.zip 用 unzip 解压



注:在make的过程中,需要安装大量的依赖库,本人因为其中一个依赖库未装成功,重装了系统,结果还是因为这个库失败…折腾了大半天,好在最后成功安装了,泪目/(ㄒoㄒ)/~~
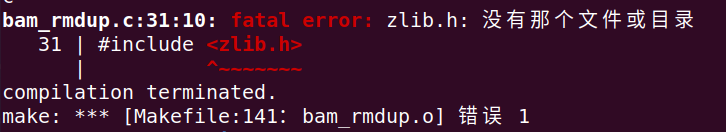
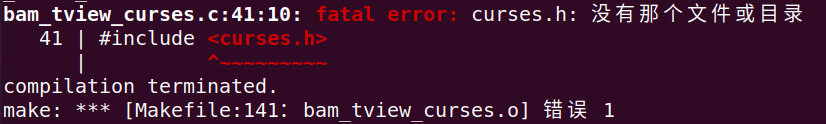

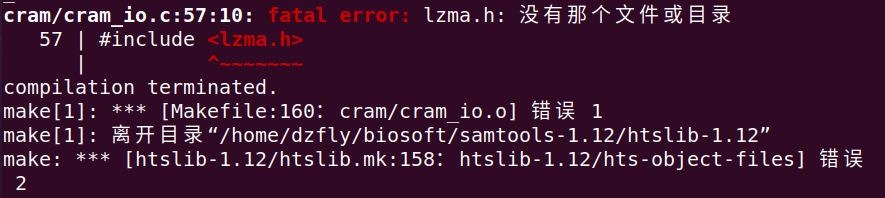

# 解决方法,根据图的顺序进行输入
sudo apt-get install zlib1g-dev
sudo apt-get install libncurses5-dev
sudo apt-get install libbz2-dev
sudo apt-get install -y liblzma-dev
sudo apt-get install libcurl4-gnutls-dev
12. 打开后缀为BAM的文件,找到产生该文件的命令。
# 进入bam文件所在的文件夹
cd /home/dzfly/biosoft/rmDuplicate/samtools/single
# 在tmp.header文件中查看生成bam文件的命令
# 只看后三行
tail -n 3 tmp.header
## 使用samtools查看bam文件
# 未将samtools加入环境变量,故使用时需敲出完整路径
~/biosoft/samtools-1.12/samtools view tmp.rmdup.bam
## 将samtools加入环境变量
# 打开环境变量配置
vim ~/.bashrc
# 添加如下语句,wq保存并退出
export PATH="$PATH:/home/dzfly/biosoft/samtools-1.12/"
# 更新环境变量
source ~/.bashrc
# 上方使用samtools查看bam文件可改为
samtools view tmp.rmdup.bam
产生bam文件的命令:
/home/jianmingzeng/biosoft/bowtie/bowtie2-2.2.9/bowtie2-align-s --wrapper basic-0 -p 20 -x /home/jianmingzeng/reference/index/bowtie/hg38 -S /home/jianmingzeng/data/public/allMouse/alignment/WT_rep2_Input.sam -U /tmp/41440.unp


13. 根据上面的命令,找到我使用的参考基因组 /home/jianmingzeng/reference/index/bowtie/hg38 具体有多少条染色体。
在tmp.header中的“SN:chr**”对应就是参考基因库的染色体条数
#使用正则表达式进行匹配
# -o 匹配
# -E 使用 | 来分割多个pattern,以此实现OR操作
# 0-9 a-z A-Z 匹配所有的数字、字母
# + 表示查找的内容在目标对象中连续出现一次或多次
# sort uniq 排序并去重
# wc -l 统计行数
grep -o -E "SN:chr[0-9]+|SN:chr[a-zA-Z]+" tmp.header |sort |uniq |wc -l
# 或者
grep -o -E "SN:chr[0-9]{1,2}|SN:chr[a-zA-Z]{1,2}" tmp.header |sort |uniq |wc -l

14. 上面的后缀为BAM 的文件的第二列,只有0和16两个数字,用 cut/sort/uniq等命令统计它们的个数。
# cut -f 2 只取第二列
# sort -n 按照数值的大小排序
# uniq -c或--count 在每列旁边显示该行重复出现的次数。
# uniq -d或--repeated 仅显示重复出现的行列。
samtools view tmp.rmdup.bam |cut -f 2 |sort -n |uniq -dc
0出现了16次,16出现了12次

15. 重新打开 rmDuplicate/samtools/paired 文件夹下面的后缀为BAM 的文件,再次查看第二列,并且统计
cd ..
cd paired
samtools view tmp.rmdup.bam |cut -f 2 |sort -n |uniq -dc

16. 下载 http://www.biotrainee.com/jmzeng/sickle/sickle-results.zip 文件,并且解压,查看里面的文件夹结构
# 下载
get -c http://www.biotrainee.com/jmzeng/sickle/sickle-results.zip
# 解压
unzip sickle-results.zip
# 查看结构
tree sickle-results



17. 解压 sickle-results/single_tmp_fastqc.zip 文件,并且进入解压后的文件夹,找到 fastqc_data.txt 文件,并且搜索该文本文件以 >>开头的有多少行?
cd sickle-results/
unzip single_tmp_fastqc.zip
cd single_tmp_fastqc/
# '^':指匹配的字符串在行首
# '$':指匹配的字符串在行尾
# '\':转义字符
grep ^\>\> fastqc_data.txt | cat -n


18. 下载 http://www.biotrainee.com/jmzeng/tmp/hg38.tss 文件,去NCBI找到TP53/BRCA1等自己感兴趣的基因对应的 refseq数据库ID,然后找到它们的hg38.tss 文件的哪一行。
wget -c http://www.biotrainee.com/jmzeng/tmp/hg38.tss
# 去指定网站查找ID
# https://www.ncbi.nlm.nih.gov/gene/7157
# 在文件中查找
grep NM_001126112 hg38.tss -n
grep NM_001126113 hg38.tss -n


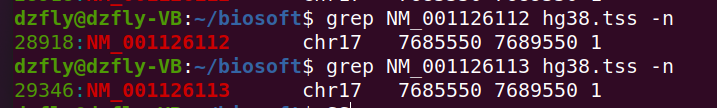
19. 解析hg38.tss 文件,统计每条染色体的基因个数。
cat hg38.tss |cut -f 2 |grep -o -E "chr[0-9]{1,2}|chr[a-zA-Z]{1,2}" |sort |uniq -dc
#或者
cat hg38.tss |cut -f 2 |grep -o -E "chr[0-9]+|chr[a-zA-Z]+" |sort |uniq -dc

20. 解析hg38.tss 文件,统计NM和NR开头的序列,了解NM和NR开头的含义。
grep -o -E "NM|NR" hg38.tss |sort |uniq -dc

NM:mRNA mixed,转录组产物序列;成熟mRNA转录本序列
NR:RNA mixed,非编码的转录子序列,包括结构RNAs,假基因转子等