
大家好,我是粥粥。
LAMMPS 是一种经典的分子动力学代码,专注于材料建模。它是大型原子/分子大规模并行模拟器的首字母缩略词。LAMMPS 具有固态材料(金属、半导体)和软物质(生物分子、聚合物)以及粗粒或中等系统的势函数。它可用于模拟原子,或者更笼统地说,作为原子、细观或连续尺度上的并行粒子模拟器。
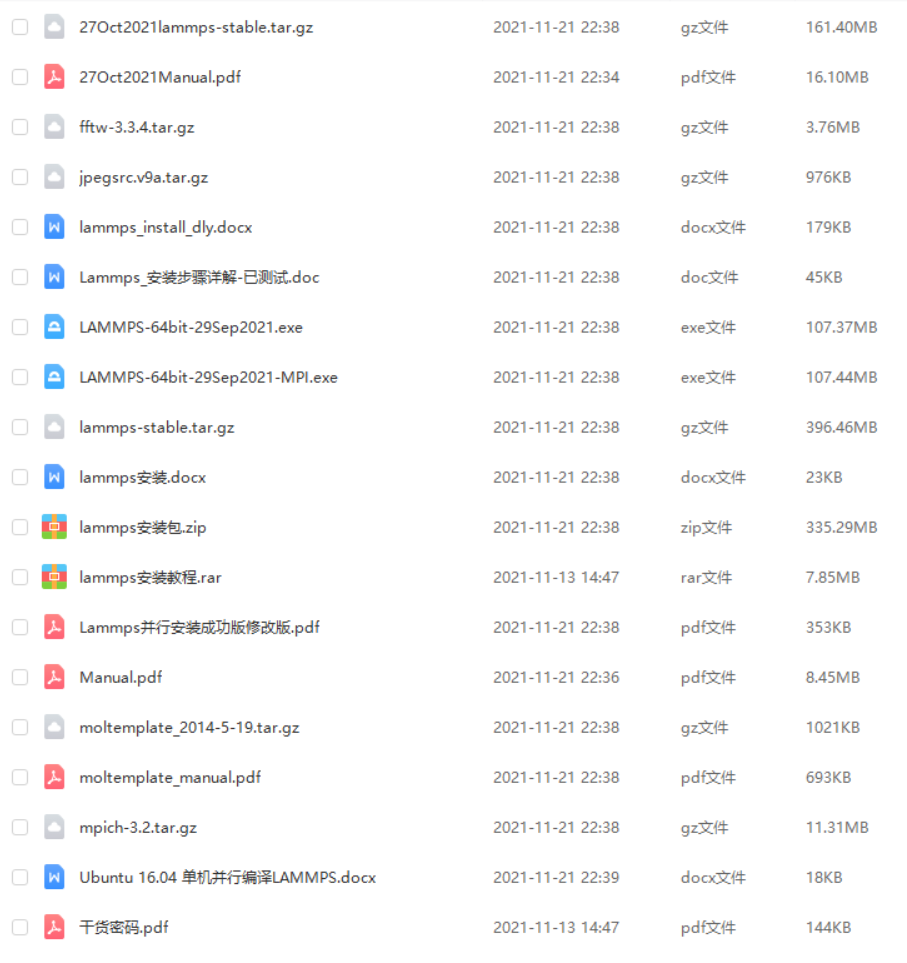
LAMMPS软件分为WIN版和LINUX版,本文提供WIN版的LAMMPS软件、LINUX版的LAMMPS软件和必要的环境包,如FFTW、MPICH。具体安装教程可在bilibili搜索《Lammps最新详细安装教程》。另外,也提供了最新版的LAMMPS官方手册PDF版。
软件安装包
WIN版的LAMMPS软件
LINUX版的LAMMPS软件
公众号内关键词回复获取:LAMMPS软件包
References
参考资料
https://www.lammps.org/
https://www.bilibili.com/video/BV12X4y1G71t?spm_id_from=333.999.0.0
如果您觉得有用的话,请点赞关注一下吧!抱拳啦!
答